
The world’s chronic disease, chronic obstructive pulmonary disease (COPD), is among the most incredibly morbid and mortal. A relatively common, preventable and curable condition, COPD most often reflects itself in chronic breathing problems and airway obstruction. Its cause is elusive and not well understood. The most important pathological processes involve oxidative stress, protease dysfunction, mitochondrial dysfunction and airway inflammation. Drug and oxygen treatment are the two main drugs used for COPD today to treat dyspnea, sputum, asthma and chest tightness. But there are also a great number of side effects to the drugs in clinical COPD prevention and treatment. Currently, the medications to prevent and treat COPD are still very limited, and there’s no treatment to stop it from worsening. A series of recent experiments suggests that inhibition of the cyclic adenosine monophosphate (cAMP) pathway has therapeutic potential for respiratory diseases. cAMP is one of those second messengers that is all around us. It is principally involved in controlling material metabolism within cells, and mainly relaxes airway smooth muscle cells and inhibits inflammation. Boosting intracellular cAMP can ameliorate lung disorders including asthma, COPD and idiopathic pulmonary fibrosis. By activating ACs or inhibiting PDEs, intracellular cAMP can be raised to ameliorate lung function, a good prevention and therapy for COPD. This article mainly describes the intracellular proteases that control cAMP, the process of cAMP regulation to avoid and cure COPD, and associated drugs, to offer an overview for the creation of COPD-related drug based on the cAMP pathway.
What is cAMP?
It was identified in 1957 and is an information molecule that is made after the first messenger attaches itself to the target cell. It’s involved in all manner of biological processes: hormone release, glycogen-processing, smooth-muscle relaxation, heart beat, learning, memory, etc. The standard cAMP signalling system is extracellular agonists bind to G protein-coupled receptors (GPCRs) and alter their conformation. Gs protein activates ACs and activates AC to oxidise adenine triphosphate (ATP) to produce cAMP. cAMP’s intracellular rise will also activate protein kinase A (PKA) and exchange protein (Epac) directly induced by cAMP, signaling to direct downstream reactions. GPCRs, ACs and PDE actively manage cAMP signaling. Intracellular cAMP level is modulated by PDE, which turns cAMP into 5′-adenine nucleotides (5’-AMP) and stops its signal transduction. Further, AKAP keeps cellular cAMP compartmentalised through the creation of a complex, which regulates certain cellular effects. Already in preliminary work, cAMP once raised also activated PKA and Epac, which in turn induced effectors on various channels, resulting in relaxation of airway smooth muscle, inhibition of airway smooth muscle growth, and modulation of cytokine production.
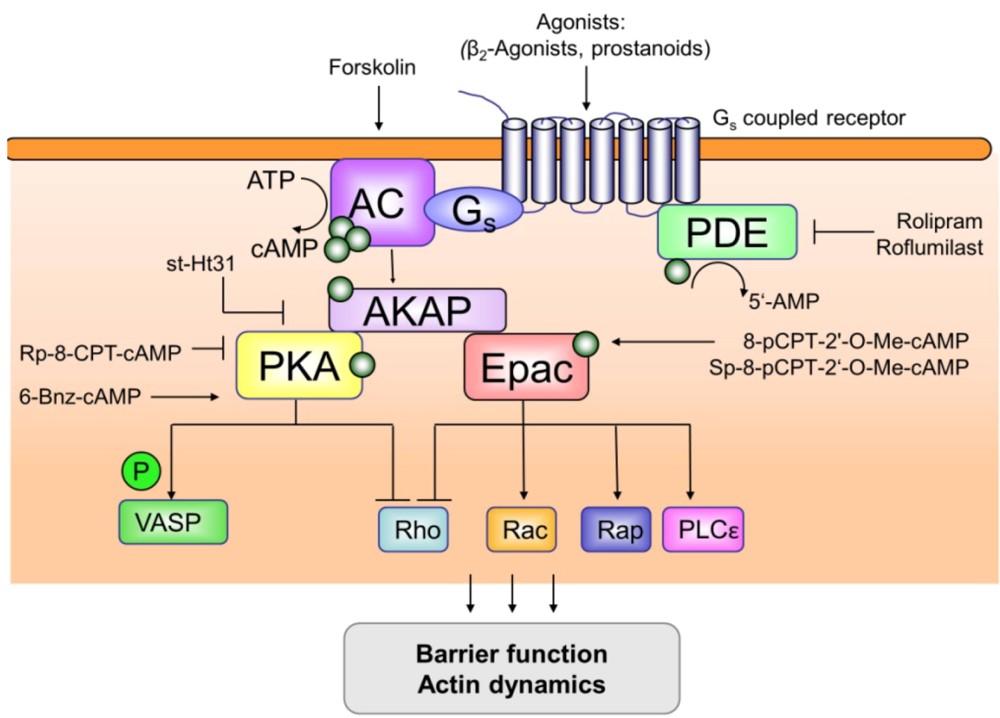
Figure 1. Overview of compartmentalization of cAMP signaling.
Proteases that Regulate cAMP
PDEs
PDEs can hydrolyse cAMP and cyclic guanosine monophosphate. We now recognise 11 isoenzyme-containing PDE subtypes (PDE1-PDE11) in mammals. PDE4 is the main cAMP hydrolyser among them. There are four subtypes of PDE4: PDE4A, PDE4B, PDE4C and PDE4D. It’s abundantly present in tissues and plays roles in physiology: the modulation of inflammation, microvascular permeability and fibrosis, contraction and expansion of smooth muscles of the bloodstream and airways. The negative correlation between PDE4 and cAMP is the primary driver of several pathological and physiological conditions including airway inflammation, COPD, asthma, psoriasis and rheumatoid arthritis. PDE4 is mostly found in lung structural cells (smooth muscle cells, airway epithelial cells, inflammatory cells) that maintain and relax airway smooth muscles and engage in a broad range of cell-life processes.
ACs
In 1989, the first mammalian AC was discovered in the bovine brain and purified and cloned. Subsequently, 9 AC subtypes were discovered. Currently, it has been determined that there are 10 subtypes of mammalian AC, 9 transmembrane AC subtypes (mAC) and one soluble AC (sAC). As a soluble enzyme, sAC is different from other AC subtypes in mammals. sAC is not regulated by G protein. The stimulation of calcium and bicarbonate is the main factor affecting it. It is mainly related to sperm motility and the growth of neuronal processes. mAb consists of a nitrogen terminus (N-terminus), two transmembrane domains, which are connected by C1a and C1b regions, and connected to the carbon terminus (C-terminus) by C2a and C2b in the cell. The N-terminus and C-terminus extend into the cytoplasm. C1 and C2 are two highly homologous cytoplasmic regions, which are the catalytic domains of AC and affect the activity of AC. The sequence and length of the N-terminus are significantly different in different ACs. Most tissues and many cell types express more than one AC subtype, and the expression of each subtype is inconsistent between different species. AC1 is highly expressed in the brain, mainly affecting neuronal function, and is a suitable neuron-specific drug target for chronic pain. AC8 is mainly expressed in the brain and is believed to play a role in learning and memory; AC3 is most abundant in the olfactory epithelium and plays a role in olfaction; AC5 and AC6 are dominant in the heart and are related to cardiac contraction; AC4, AC7, and AC9 are commonly found in mammalian tissues; AC2 is more widely distributed and is expressed in the brain, lungs, skeletal muscle, and heart, and the subtypes highly expressed in the lungs are AC2, AC6, AC8, and AC9.
cAMP and COPD
Complex reaction signalling in cells is extremely controlled by cAMP. When it detects and amplifies signals, it regulates related physiological effects through cAMP-dependent enzymes or proteins. After the cells are ‘inflamed’ by the outside world, it affects the functions of AC and PDE enzymes, modulates intracellular cAMP, which affects downstream PKA, EPAC and AKAP, then nuclear factor B (NFB) and cAMP response element binding protein. (CREB), and extracellular signal-regulated kinase (ERK) activation, which leads to airway smooth muscle relaxation and diminished inflammation and airway fibrosis. Consequently, controlling the cAMP pathway is a great candidate for both prevention and cure of long-term respiratory conditions like COPD.
cAMP and Airway Smooth Muscle Relaxation
In the respiratory system of COPD patients, we see the obvious signs of airflow inhibition: wall remodeling and mucus formation. Among them, distorted growth and hypertrophy of airway smooth muscle cells (ASMCs) is one of the most important features of COPD airway remodelling. One of the most important medications for treating airflow obstruction in COPD is 2-adrenergic receptor agonists. They act as bronchodilators because intracellular cAMP levels spike. The two main effectors of cAMP, PKA and EPAc, are then mobilised and participate in smooth muscle relaxation of the airway. EPAc reverses the phenotypic conversion of airway smooth muscle with ERK, regulates contraction and relaxation of ASMCs, and skewed Ras homologous gene family member A and Ras-associated C3 botulinum toxin substrate (Rac) to Rac, to suppress myosin. light chain Phosphorylation ultimately relaxes airway smooth muscle. Another research has shown that a boost in the levels of cAMP in the smooth muscle of the airway and the bronchial epithelium can both decrease intraepithelial acetylcholine and ameliorate bronchial symptoms. Recently, it has been discovered that mutating or demethylating ATP-binding box subfamily C member 1 decreases the release of cAMP from ASMCs that is stimulated by 2 receptors, increasing intracellular cAMP, easing tension on airway smooth muscle cells and inhibiting COPD airway smooth muscle remodeling in several different ways.
cAMP and Inflammation
COPD’s root cause is chronic inflammation, whose airway inflammation is defined by increased numbers of neutrophils, alveolar macrophages and lymphocytes. The cAMP anti-inflammatory effect is varied, it regulates several biological processes, and its regulatory effect depends on the type of cell. In the COPD inflammatory process, neutrophils generate inflammatory mediators and proteases. We have found that COPD patients’ neutrophils migrate irregularly, and that they migrate to the chemoattractant leukotriene B4 (LTB4) and the chemokine CXC ligand 1 (CXCL1) with increased velocity. We also discovered that increasing the intracellular cAMP level can prevent neutrophils from chemotaxis to CXCL1 and LTB4. Bronchial macrophages, interstitial macrophages, and alveolar macrophages are the most prominent ones in the lungs; alveolar macrophages are the major players here. On alveolar macrophages, researchers have observed that macrophages spit out cytokines and chemokines after they have been activated and stimulated by CSE or other stimulants, and that cAMP induces the release of pro-inflammatory factors like tumor necrosis factor- (TNF-) and the chemokine C-C ligand 3 (CCL3), but not the anti-inflammatory cytokines like IL-10.
The other cAMP signalling mechanism in the immune system is to turn on the downstream nuclear transcription factor CREB, bind PKA to the NF-B inhibitory protein (IB) molecules to create a complex, and the signal that breaks down IB turns on the catalytic subunit of the PKA, which phosphorylates the p65 molecule. Phosphorylated CREB could block the NF-B association with the gene in question and can block gene transcription via a binding to activated NF-B to prevent inflammation.
cAMP and Epithelial-Mesenchymal Transition
And from earlier research, evidence for EMT involvement in the COPD pathogenesis is mounting. – Smoke and pollution bombard the lungs with excess oxidants (especially reactive oxygen species) which increases oxidative stress and EMT leading to a radical degeneration of COPD. Moreover, mesenchymal transition of airway, lung epithelial cells, and bronchial epithelial cells in COPD is considerably elevated. They’ve discovered that cAMP sits in particular subcellular microdomains and regulates the activity of downstream effector proteins PKA and Epac; AKAP attaches to PKA in a small -helical loop to drive EMT. It’s now even known that CSE-induced release of transforming growth factor- in Be-as-2b cells is a trigger for EMT, for the activation of type I collagen (COLI) and for the inhibition of the epithelial cell marker E-cadherin. But increasing cAMP or disrupting the direct interaction between AKAP and PKA will significantly decrease COLI upregulation, impact epithelial-mesenchymal transition in COPD, and therefore reduce COPD symptoms.
Treatment of COPD with Drugs that Balance Camp Pathway
In practice, COPD prevention and treatment medications consist mostly of inhaled glucocorticoids, long-acting 2-adrenergic receptor agonists, and long-acting M receptor antagonists, all of which are very bad. There are still few drugs available to prevent and treat COPD, and no treatment that prevents the disease from spreading. The use of drugs that regulate cAMP pathway in COPD is currently limited to PDE4 inhibitors and AC agonists.